Research
| Life Language Processing
A broad and simple definition of `language’ is a set of sequences constructed from a finite set of symbols. By this definition, biological sequences, human languages, and many sequential phenomena that exist in the world can be viewed as languages. Although this definition is simple, it includes languages employing very complicated grammars in the creation of their sequences of symbols. Examples are biophysical principles governing biological sequences (e.g., DNA, RNA, and protein sequences), as well as grammars of human languages determining the structure of clauses and sentences. This project uses a language-agnostic point of view in the processing of both biological sequences and human languages. Two main strategies are adopted toward this purpose, (i) character-level, or more accurately, subsequence-level processing of languages, which allows for simple modeling of the sequence similarities based on local information or, bag-of-subsequences, (ii) language model-based representation learning encoding contextual information of sequence elements using the neural network language models. We propose language-agnostic and subsequence-based language processing using the above-mentioned strategies in addressing three main research problems in proteomics, genomics/metagenomics, and natural languages using the same point-of-view.
|
| Protein Functional/Structural Annotation
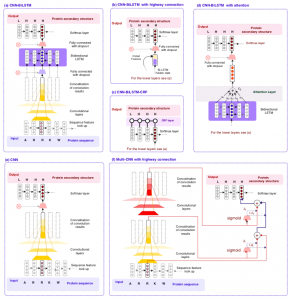
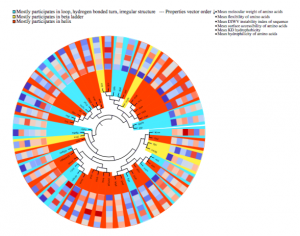
One of the main challenges in proteomics is that there exists a large gap between the number of known protein sequences and known protein structures/functions. The central question here is how to efficiently use large numbers of sequences to achieve a better performance in the structural and functional annotation of protein sequences. Here, we proposed subsequence-based representations of protein sequences and their language model-based embeddings trained over a large dataset of protein sequences, which we called protein vectors (or ProtVec). In addition, we introduced a motif discovery approach, benefiting from probabilistic segmentation of protein sequences to find functional and structural motifs. This segmentation is also inferred from large protein sequence datasets. The ProtVec approach has proved a seminal contribution in protein informatics and now is widely used for machine learning based protein structure and function annotations. We showed in different protein informatics tasks that bag-of-subsequences and protein embeddings are complementary information for language-agnostic prediction of protein structures and functions, which also achieved the state-of-the-art performance in the 2 out of 3 tasks of Critical Assessment of protein Function Annotation (CAFA) in 2018 (CAFA 3.14). Moreover, we systematically investigated the role of representation and deep learning architecture in protein secondary structure prediction from the primary sequence.
|
| Genomics/Metagenomics Phenotype and Biomarker Prediction
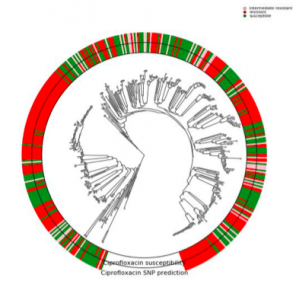
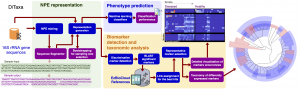
One of the prominent challenges in metagenomics involves the host phenotypic characterization based on the associated microbial samples. Microbial communities exist almost on every accessible surface on earth, supporting, regulating, and even causing unwanted conditions (e.g., diseases) to their hosts and environments. Detection of the host phenotype and the phenotype-specific taxa from the microbial samples is the chief goal here. For instance, identifying distinctive taxa for microbiome-related diseases is considered key to the establishment of diagnosis and therapy options in precision medicine and imposes high demands on the accuracy of microbiome analysis techniques. Here, we propose two distinct language-agnostic subsequence-based processing methods for machine learning on 16S rRNA sequencing, currently the most cost-effective approach for sequencing of microbial communities. We propose alignment- and reference- free methods, called MicroPheno and DiTaxa, designed for microbial phenotype and biomarker detection, respectively. MicroPheno is a k-mer based approach achieving the state-of-the-art performance in the host phenotype prediction from 16S rRNA outperforming conventional OTU features. DiTaxa, substitutes standard OTU-clustering by segmenting 16S rRNA reads into the most frequent variable-length subsequences. We compared the performance of DiTaxa to the state-of-the-art methods in phenotype and biomarker detection, using human-associated 16S rRNA samples for periodontal disease, rheumatoid arthritis, and inflammatory bowel diseases, as well as a synthetic benchmark dataset. DiTaxa performed competitively to MicroPheno (state-of-the-art approach) in phenotype prediction while outperforming the OTU-based state-of-the-art approach in finding biomarkers in both resolution and coverage evaluated over known links from literature and synthetic benchmark datasets.
|
| Multilingual and Language-Agnostic NLP on > 1000 Languages The third central problem we addressed in this project is focused on human languages. Many of 7000 world’s natural languages are low-resource and lack digitized linguistic resources. This has put many of these human languages in danger of extinction and has motivated developing methods for automatic creation of linguistic resources and linguistic knowledge for low-resource languages. To address this problem via our language-agnostic point of view (by not treating different languages differently), we develop SuperPivot for subsequence-based linguist marker detection in parallel corpora of 1000 languages, which was the first computational investigation for linguistic resource creation in such a scale. As an example, SuperPivot was used to study the typology of tense in 1000 languages. Next, we utilized SuperPivot for the creation of the largest sentiment lexicon to date in terms of the number of covered languages (1000+ languages) achieving macro-F1 over 0.75 on word sentiment prediction for most evaluated languages, meaning that we enable sentiment analysis in many low resource languages. To ensure the usability of UniSent lexica for any new domain, we propose DomDrift, a method quantifying the semantic changes of words in the sentiment lexicon in the new domain. Next, we extend the DomDrift method to quantifying the semantic changes of all words in the language. We proposed a new metric for language comparisons based on the language word embedding graphs requiring only monolingual embeddings and word mapping between languages obtained through statistical alignment in parallel corpora. We performed language comparison for fifty natural languages and twelve genetic language variations of different organisms. As a result, natural languages of the same family were clustered together. In addition, applying the same method on organisms’ genomes confirmed a high-level difference in the genetic language model of humans/animals versus plants. This method called word embedding language divergence is a step toward unsupervised or minimally supervised comparison of languages in their broad definition. 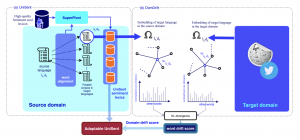  |