DeepPrime2Sec: Deep Learning for Protein Secondary Structure Prediction from the Primary Sequences
 |
DeepPrime2Sec
DeepPrime2Sec and the used datasets are available here under the Apache 2 license. |
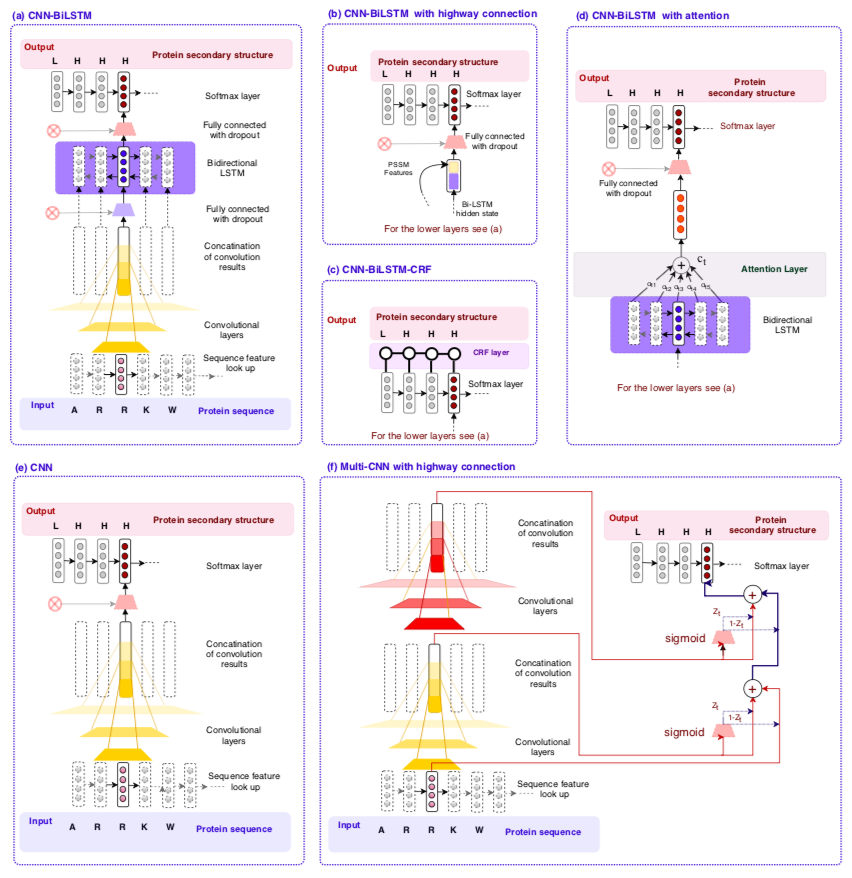
Here we investigate deep learning-based prediction of protein secondary structure from the protein primary sequence. We study the function of different features in this task, including one-hot vectors, biophysical features, protein sequence embedding (ProtVec), deep contextualized embedding (known as ELMo), and the Position Specific Scoring Matrix (PSSM). In addition to the role of features, we evaluate various deep learning architectures including the following models/mechanisms and certain combinations: Bidirectional Long Short-Term Memory (BiLSTM), convolutional neural network (CNN), highway connections, attention mechanism, recurrent neural random fields, and gated multi-scale CNN. Our results suggest that PSSM concatenated to one-hot vectors are the most important features for the task of secondary structure prediction.
Utilizing the CNN-BiLSTM network, we achieved an accuracy of %69.9 and %70.4 using ensemble top-k models, for 8-class of protein secondary structure on the CB513 dataset, the most challenging dataset for protein secondary structure prediction. Through error analysis on the best performing model, we showed that the misclassification is significantly more common at positions that undergo secondary structure transitions, which is most likely due to the inaccurate assignments of the secondary structure at the boundary regions. Notably, when ignoring amino acids at secondary structure transitions in the evaluation, the accuracy increases to %90.3. Furthermore, the best performing model mostly mistook similar structures for one another, indicating that the deep learning model inferred high-level information on the secondary structure.
The DeepPrime2Sec implementation allows to investigate the role of different features and deep learning architecture in the task of protein secondary structure. New architectures can be easily added to the software.